1. Let’s Recap¶
We are interested in the (ground-state) solutions of the molecular electronic Schrödinger equation (i.e. under the Born–Oppenheimer approximation).
So far we have seen:
- Hartree–Fock (HF): A mean-field approximation in which the wave-function is written as a single Salter determinant. The molecular orbitals (MO) used in the HF wave-function are expressed as a linear combination of basis functions (usually atomic orbitals). The coefficients of the MO expansion are obtained variationally and are the only wave-function parameters.
- Full Configuration Interaction (FCI): The exact solution to the electronic Schrödinger equation in a given one-electron basis. The FCI wave-function is expressed as a linear combination of all possible Slater determinant. The coefficients of the FCI expansion are obtained variationally and are the only wave-function parameters.
- Truncated CI and Møller–Plesset (MP) theory: Post HF models providing intermediate results between HF and FCI (compromise between accuracy and computational cost).
- More things that are not really relevant here…
In the wave-function based models described above and in Coupled Cluster (CC) theory as well, we make approximations of two different kinds:
- In the one-electron space (choice of a finite basis set).
- In the N-electron space (choice of a wave-function model).
In this lecture we are only concerned with the approximations introduced in the N-electron space. We will therefore always consider solutions to the electronic Schrödinger equation for a particular choice of one-electron basis set, keeping in mind that for approaching the exact solution it is important to make improvement in both spaces. A FCI solution for a minimal basis set is not very useful in practice…
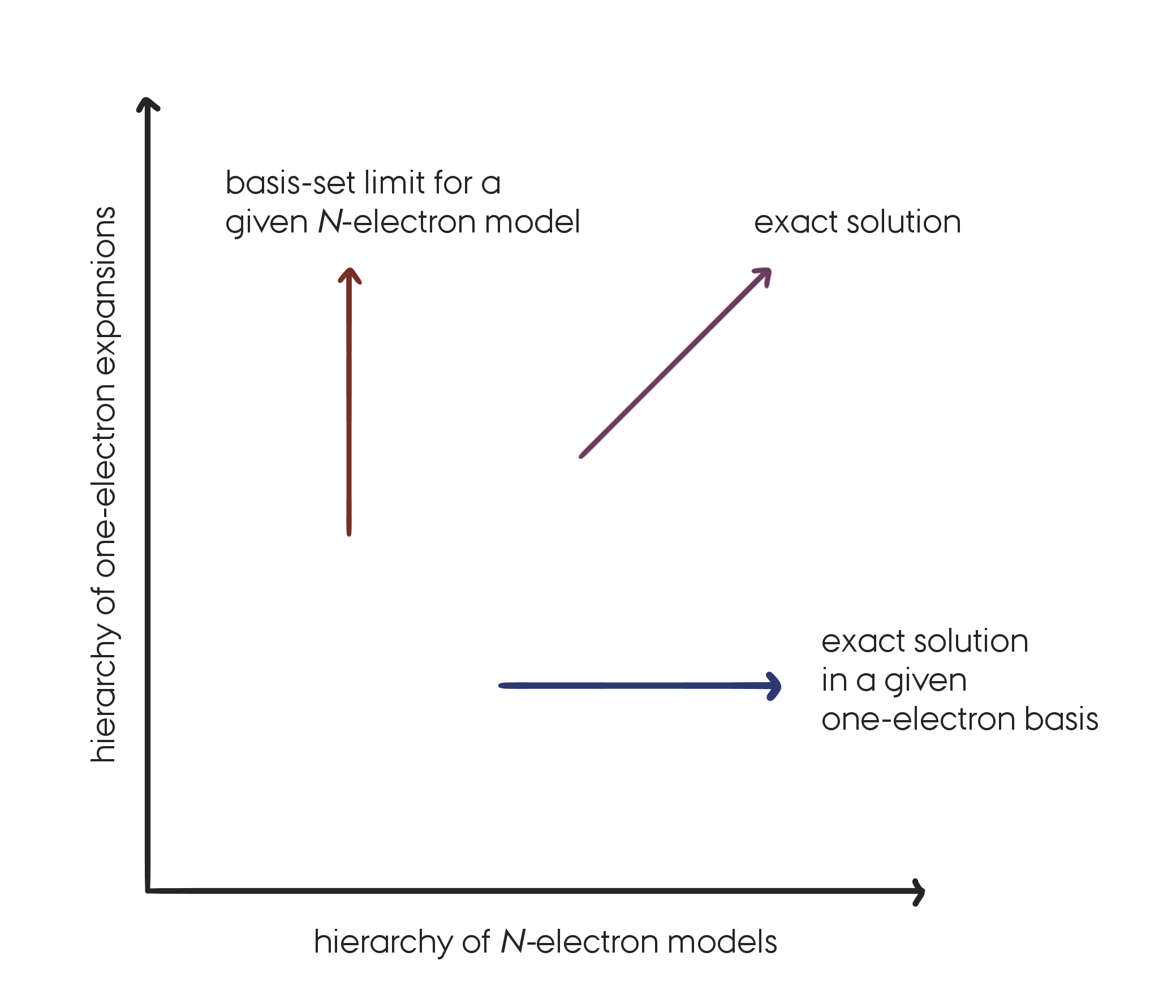
The systematic approach to the exact solution of the Schrödinger equation by successive improvements in the one- and N-electron spaces. (Replicated from Figure 5.1 in [Helgaker2000]).